Enhancing single-cell transcriptomics using interposed anchor oligonucleotide sequences
Video
An anchor-interposed bead is designed to enhance single-cell long-read transcriptomics by simply placing an anchor sequence between cell barcode and a UMI. This anchor provides a clear demarcation point, enhancing UMI recognition and minimising synthesis errors.
NDORMS News
News about this research was published on NDORMS News, check here
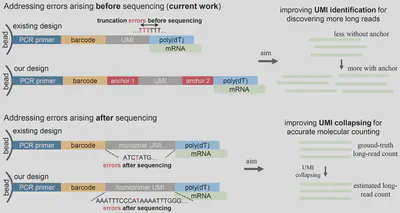
To better convey the core contributions of our work to a broader audience, I am here trying to clarify the fundamental distinction between two major studies we published over the past year. One study focuses on eliminating PCR artefacts to improve the accuracy of molecular quantification, through what we call the homotrimer UMI demultiplexing approach. The other, which is the subject of our current work, addresses the issue of truncated UMIs caused by poly(T) sequences, thereby enhancing the efficiency of capturing long-read sequences.
Undoubtedly, both studies aim to improve sequencing accuracy and involve both experimental procedures and computational analysis. In both cases, nucleotide sequences on beads were specifically modified during the experimental stage, followed by downstream computational analysis after sequencing. However, the key difference lies in the source of the errors each method addresses. The anchor bead approach in our current study tackles errors that are inherent to the bead itself before sequencing, whereas the homotrimer UMI demultiplexing approach corrects errors introduced during sequencing. Accordingly, we classify these two technologies as prior to sequencing and post-sequencing error correction methods, respectively. This distinction is reflected in the “prior to sequencing” and “post-sequencing” labels in the figure published on the NDORMS news.
Looking deeper, PCR artefacts are largely unavoidable given the current limitations of experimental platforms and techniques. Therefore, modifying oligonucleotide sequences before sequencing, as done in the homotrimer UMI approach, does not directly impact molecular quantification accuracy during this stage. Error correction must therefore be performed post-sequencing. In contrast, the anchor bead strategy mitigates UMI misidentification by minimising errors from the bead itself during the sequencing process, through sequence modification performed before sequencing begins.
Publication
Please check our paper here in Communications Biology .
1. Aim
This study aims to address UMI Truncation and PolyT Contamination in Single-Cell Long-Read Sequencing.
2. Background
To investigate the prevalence and origin of UMI truncation in single-cell RNA-sequencing (scRNA-seq), we conducted a large-scale analysis using publicly available short-read datasets from 10x Genomics. By examining the nucleotide composition at the terminal end of UMI sequences, we observed a statistically abnormal enrichment of thymine (T) bases. This phenomenon is inconsistent with random nucleotide distribution and strongly suggests contamination of the UMI tail likely due to polyT extension into the UMI region. Such contamination results in reduced UMI sequence diversity and undermines accurate molecular counting.
3. Problem
This observation raises a critical question: is the polyT enrichment at the UMI tail due to:
Correct identification of the UMI start site but a UMI that naturally ends with many T bases?
Incorrect identification of the UMI boundary, causing polyT sequences from downstream regions to be misincluded in the UMI?
While it is difficult to definitively determine which extracted UMI is “correct” based solely on its terminal composition, our hypothesis is that if we accurately locate the start of the UMI, we at least eliminate upstream ambiguity. The remaining T content can then be evaluated post hoc.
4. Solution (anchor for UMI boundary recognition)
To address this challenge, we introduced a short 4-nucleotide anchor sequence (BAGC) between the cell barcode and the UMI in the bead design. This anchor serves as a unique positional marker that allows unambiguous identification of the UMI start site in long-read sequencing data. This design was incorporated to the scCOLOR-seq protocol as scCOLOR-seqv2.
We evaluated the base composition of extracted UMIs from reads sequenced with scCOLOR-seqv2. We observed a significant reduction in terminal T-content, strongly suggesting that anchor-guided UMI extraction minimises polyT contamination. In contrast, a control approach that extracts the UMI by counting a fixed number of bases downstream from a known primer position failed to eliminate polyT bias likely due to the influence of indels that shift the effective start site of the UMI. While we did not experimentally validate the role of indels in this context, the data suggest a strong possibility that such structural variations contribute to incorrect UMI boundary identification when positional methods are used.
To further assess the robustness of the anchor approach under variable sequencing conditions, we conducted in silico simulations mimicking realistic long-read sequencing scenarios. These simulations introduced indel and substitution errors during both the PCR amplification and sequencing phases.
5. Computational simulation report
You can find how we made this simulation here .
Across a range of error rates, the anchor-based method consistently outperformed positional extraction, significantly improving the proportion of long reads in which the UMI could be successfully and accurately recovered. This improvement is critical for downstream applications such as gene expression quantification, especially in single-cell contexts where data is sparse and sensitive to noise.
6. Implications and significance
Sequencing remains an error-prone process. Accurately revealing biology, especially in the context of transcript quantification, requires not only advancements in laboratory methods but also the design of computational strategies that account for and correct technical artifacts.
7. Concluding remarks
Our findings show that eliminating polyT-induced UMI truncation through anchor-guided UMI extraction significantly increases the proportion of usable long reads in single-cell datasets. This is especially valuable in studies of rare genetic diseases, where the expression of key genes may be low or difficult to detect. By maximising the recovery of informative reads, this anchor bead enhances the resolution and accuracy of single-cell transcriptomic analysis.