Concept#
TMKit contains seqNetRR, a high-performance computing library for constructing residue connections in sequence and contact-map context and performing the assignment of co-evolutionary features (or other types of co-variant features) in linear time. This module is aimed at facilitating co-evolutionary feature engineering, scheme design, and organization for machine learning modelling of interactions/contacts between residues.
In this tutorial, we will go through the definition of global residue-residue connections (GlobRRCs) and the computational scheme to construct them at a fast speed.
GlobRRCs: global residue–residue connections (GlobRRCs) are those formed by one residue of a residue pair of interest (including its neighbors) and the serially ordered neighboring residues of the other one residue of the residue pair (including itself, e.g. connections between residue 1 and the neighboring residues of residue 2).
Assigning features to co-variant residue pairs in transmembrane proteins is notoriously challenging due to their long sequences, which generate a substantial number of residue pairs, making the process computationally intensive. The seqNetRR library significantly reduces runtime by implementing modular methods to minimise the overhead of frequent Python function calls and leveraging hash tables for efficient data retrieval and assignment. Benchmarking against Pandas and NumPy confirms that seqNetRR is the fastest method for assigning co-evolutionary features to GlobRRCs, offering an optimized solution for large-scale transmembrane protein analysis.
Problem analysis#
Suppose we have a protein sequence of length L. The total number of residue pairs N extracted from the sequence is given by
All residue pairs can be represented as a 2D array or a 2D data object in Python. When a sliding window of size w is applied, the structure expands into a 3D data object, as illustrated in the plot below.
Constrained by t edges in a bipartite graph, the number of window-placed GlobRRCs Nt for each residue is calculated as
leading to a total of 2×Nt GlobRRCs for each residue pair.
The core challenge is efficiently assigning features to this large volume of residue pairs. A naive approach would result in an O(n³) time complexity, making it computationally infeasible for large datasets. However, by leveraging hashing techniques and optimizing the process to eliminate unnecessary computations, we can dramatically reduce runtime. Among various methods tested, the Hash-based approach has proven to be the most efficient.
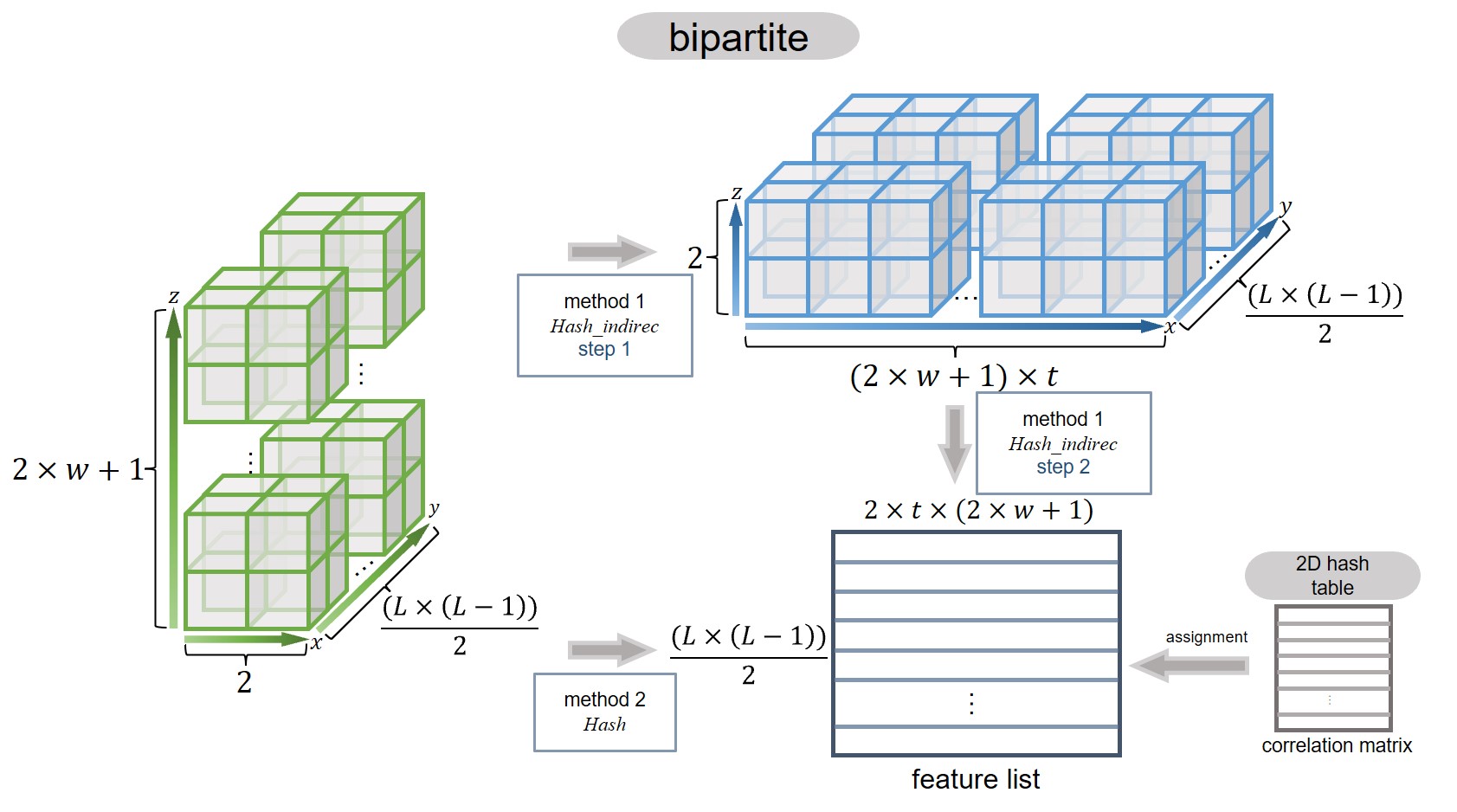
Caption: Computing scheme of assigning features to GlobRRCs in seqNetRR.#
Method |
Description |
|---|---|
|
achieved with method 2 by assigning features and in the meantime constructing the entire list of residue pairs. Feature assignment is done by querying values from a hash table. |
|
achieved with method 1 by constructing the entire combinations of residues first and then going to feature assignment. |
|
achieved with method 2 by assigning features and in the meantime constructing the entire list of residue pairs. Feature assignment is done by pandas.at. |
|
achieved with method 2 by assigning features and in the meantime constructing the entire list of residue pairs. Feature assignment is done by numpy array. |
Runtime evaluation#
Tip
TRAIN, PREVIOUS, and TEST used in DeepHelicon[1] are used as benchmark datasets to gauge the runtime and downloaded via this link
Computing performance is gauged by the running time of different methods. We first evaluated the running time of different methods on assigning co-evolutionary features according to GlobRRCs restricted by a bipartite graph, a 5×5 patch (refer to here). We excluded the comparison with the NumPy array indexing method as we found it extremely time-consuming.
As illustrated in the plot below, methods utilizing hash tables significantly outperform the pandas.at method in terms of average runtime efficiency. Notably, the Hash method achieves an execution time of 6.47s (6.60s for TRAIN, 7.48s for PREVIOUS, and 5.33s for TEST). Paired t-tests confirm that the runtime differences between all method pairs are statistically significant. Additionally, a comparative analysis between Hash_indirec and Hash underscores that optimizing performance requires eliminating even the smallest unnecessary computational costs.
Furthermore, the runtime per molecule exhibits a strong exponential correlation with molecular length and a linear correlation with the number of residue pairs, as evidenced by exceptionally high R-squared values (0.95 and 1.00, respectively). For instance, for a protein of length 500 with approximately 120,000 non-redundant residue pairs, seqNetRR efficiently isolates GlobRRCs from other connections in the residue contact network, completing feature assignment in ~16 seconds.
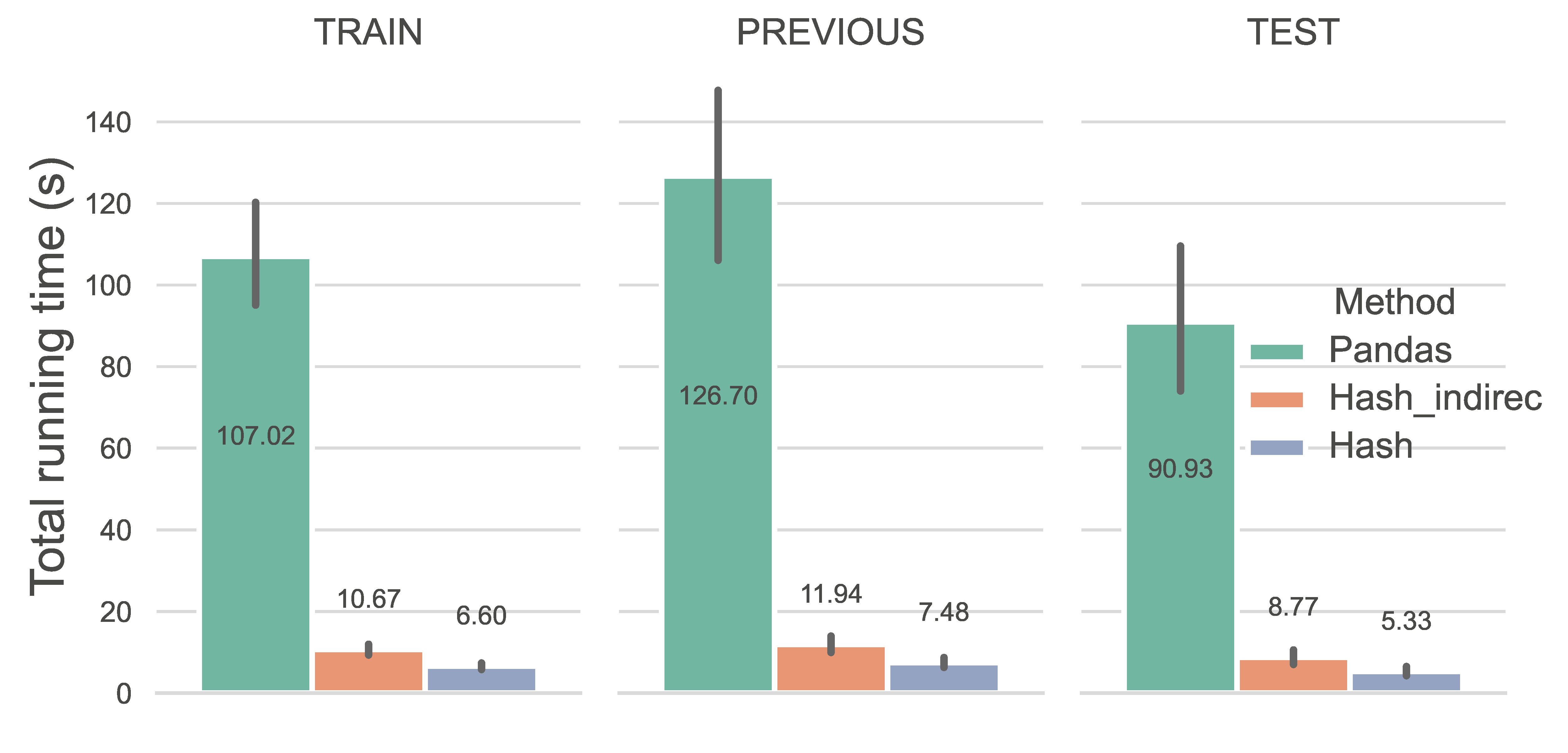
Caption: The runtime of the two tasks at the per-protein level.#