Protein-ligand binding pocket#
Identification of protein-ligand binding pockets is crucial for understanding many biological processes that is mediated by ligands and its visualization can help drug discovery and biological interpretation. TMKit allows users to show the pocket details of a protein-ligand complex.
Reminder of data
Please make sure that the build-in example dataset has been downloaded before you walk through the tutorial.
Example usage#
The protein structure 6feq is a transmembrane protein-ligand complex, showing a structure of inhibitor-bound human multidrug transporter ABCG2 (as shown in the plot below). A small molecule inhibitor (or ligand) Ko143 can inhibition the biological functions of ABCG2. We extract the complex structure and show the protein-ligand pocket in multiple forms . The complex has been placed in data/example/vs/.
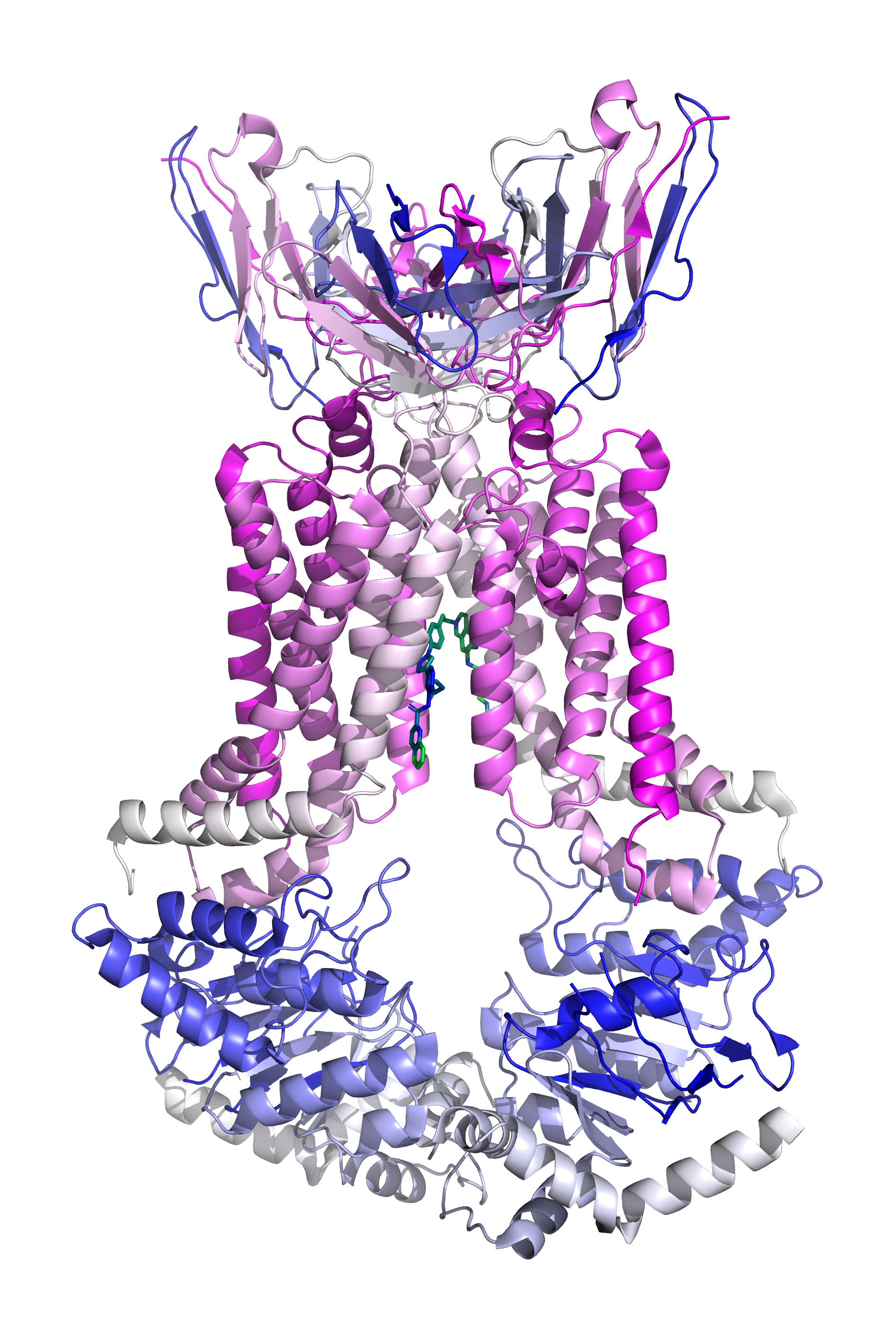
Caption: Protein-ligand complex of 6feq#
First, we can visualize it in rainbow form annotated with residues around the ligand using the following code.
import tmkit as tmk
tmk.vs.sm_local(
prot_name='6feq',
pdb_complex_fp=to('data/example/vs/'),
sm_rep="sticks",
nby_rep='sticks',
prot_c='blue_white_magenta',
sm_c='blue_green',
pocket_rep='stick'
)
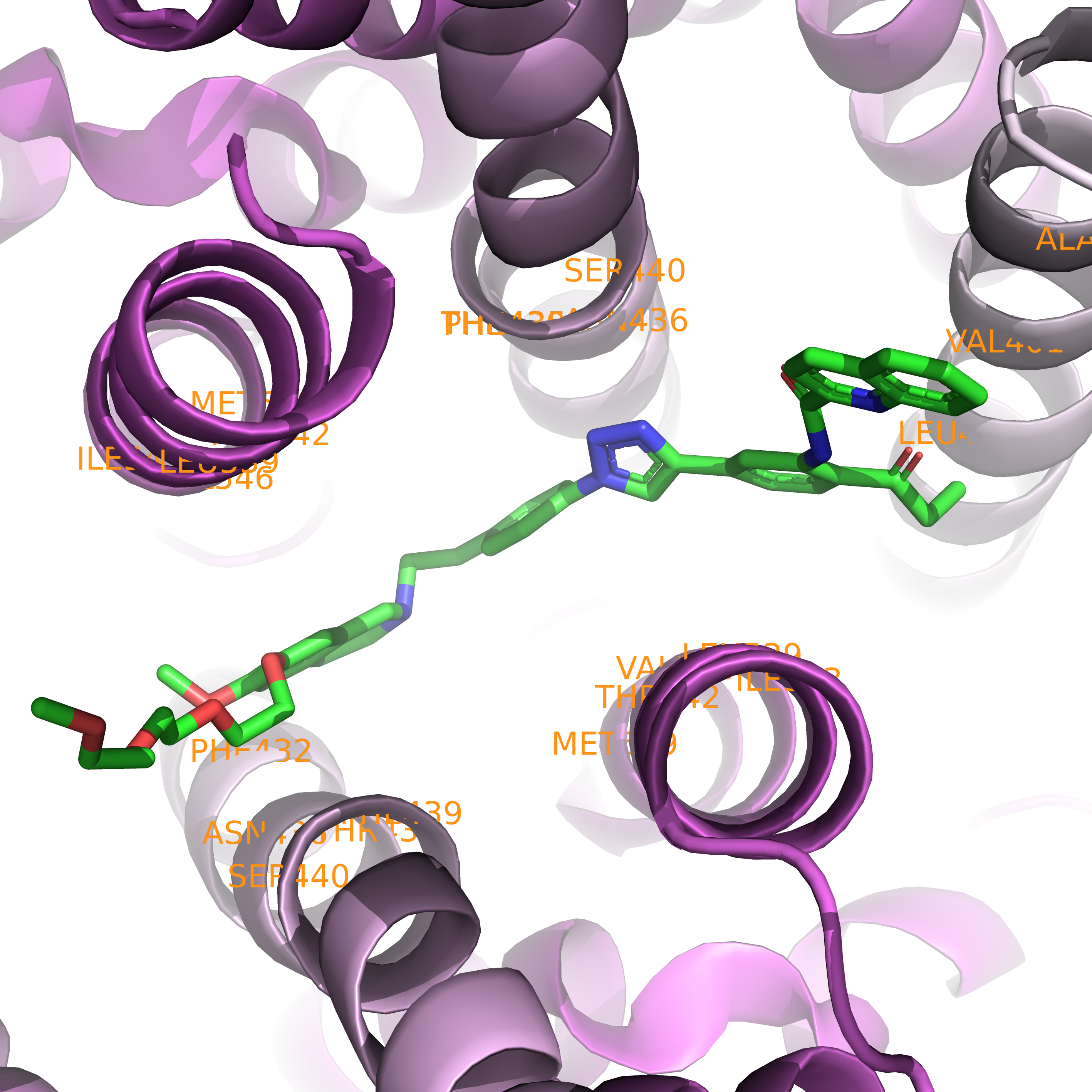
Caption: Close-up of the binding pocket in rainbow form.#
Second, we can visualize it in sphere form with no residue annotation using the following code.
import tmkit as tmk
tmk.vs.sm_local(
prot_name='6feq',
pdb_complex_fp='data/pdb/rcsb/',
sm_rep="sticks",
nby_rep='sticks',
prot_c='blue_white_magenta',
sm_c='blue_green',
pocket_rep='sphere'
)
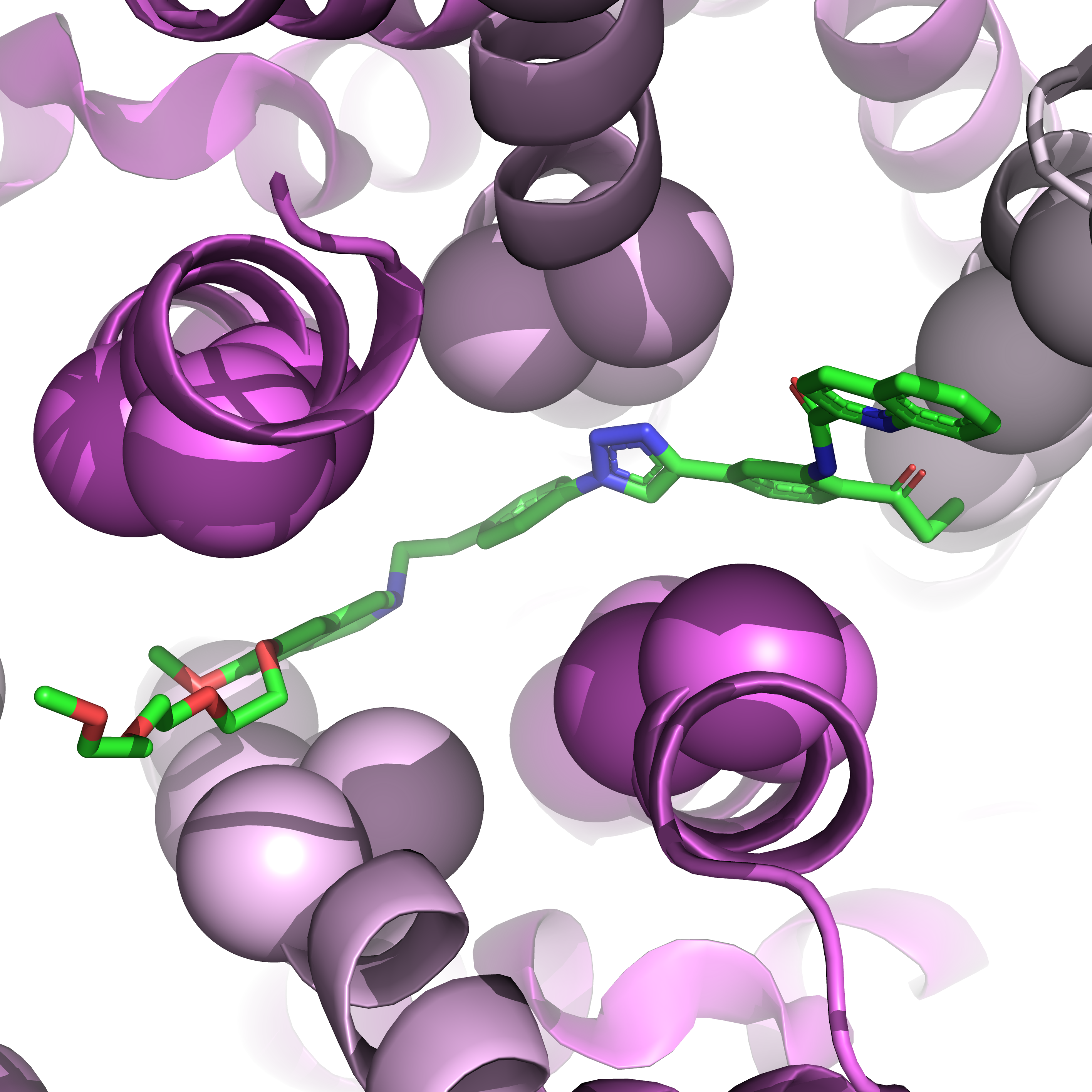
Caption: Close-up of the binding pocket in sphere form.#
Third, we can visualize it in surface form with no residue annotation using the following code.
import tmkit as tmk
tmk.vs.sm_local(
prot_name='6feq',
pdb_complex_fp='data/pdb/rcsb/',
sm_rep="sticks",
nby_rep='sticks',
prot_c='blue_white_magenta',
sm_c='blue_green',
pocket_rep='surface'
)
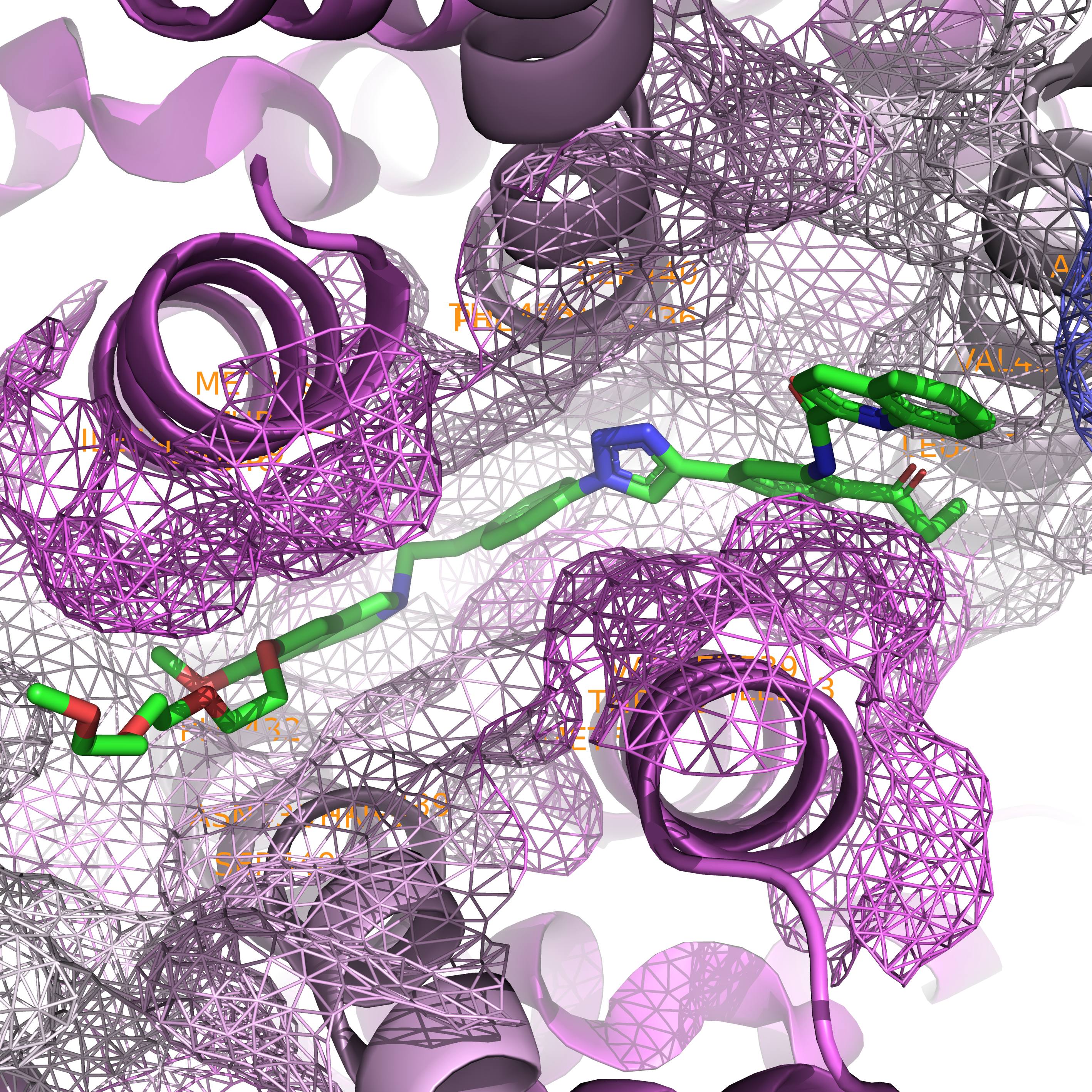
Caption: Close-up of the binding pocket in surface form.#
Attributes#
Attribute |
Description |
|---|---|
|
name of a protein in the prefix of a PDB file name (e.g., 1xqf in 1xqfA.pdb) |
|
path where a target protein complex file is place |
|
color of the entire protein |
|
representation of a ligand |
|
representation of amino acid residues surrounding a ligand |
|
color of a ligand |
See also
Please see here for better understanding the file-naming system.
Output#
Finally, you will see the following output.
PyMOL(TM) Molecular Graphics System, Version 2.5.0.
Copyright (c) Schrodinger, LLC.
All Rights Reserved.
Created by Warren L. DeLano, Ph.D.
PyMOL is user-supported open-source software. Although some versions
are freely available, PyMOL is not in the public domain.
If PyMOL is helpful in your work or study, then please volunteer
support for our ongoing efforts to create open and affordable scientific
software by purchasing a PyMOL Maintenance and/or Support subscription.
More information can be found at "http://www.pymol.org".
Enter "help" for a list of commands.
Enter "help command-name" for information on a specific command.
Hit ESC anytime to toggle between text and graphics.
Detected OpenGL version 4.6. Shaders available.
Detected GLSL version 4.60.
OpenGL graphics engine:
GL_VENDOR: NVIDIA Corporation
GL_RENDERER: NVIDIA GeForce RTX 2070/PCIe/SSE2
GL_VERSION: 4.6.0 NVIDIA 536.40